Table Of Content
- How did your time at the USC Mann School impact you?
- Drug resistance mutation analysis
- Synergy Between the Two Approaches¶
- Pharmaceutical Sciences (PSCI)
- TransformerCPI2.0: predicting compound protein interaction without using protein structure
- PocketFlow is a data-and-knowledge-driven structure-based molecular generative model
- Molecules generated with DRAGONFLY potently and selectively activate PPARγ
- Myeloid cell headwinds for GD2-CAR-T cell therapy
Other cutting edge, state-of-the-art tools provided by the center include the 600 MHz Nuclear Magnetic Resonance facility, the Agilent Liquid chromatography mass spectrometry, several high-performance liquid chromatographs, and most recently the in-house pharmacokinetic laboratory. The center is currently offering a course on HIV Drug Discovery covering the topics of HIV Replication Cycle, Target Selection, Nucleoside and Nucleotide Reverse Transcriptase Inhibitors, Entry Inhibitors, and Intellectual Property in Drug Discovery. No GRE scores are required, and students may enroll in classes before applying for the Certificate if desired.
How did your time at the USC Mann School impact you?
The selection of the graph-to-sequence architecture was made to facilitate the development of deep learning models capable of supporting both ligand-based and structure-based molecular design. Drug development based on target proteins has been a successful approach in recent decades. However, the conventional structure-based drug design (SBDD) pipeline is a complex, human-engineered process with multiple independently optimized steps. Here, we propose a sequence-to-drug concept for computational drug design based on protein sequence information by end-to-end differentiable learning. First, we design TransformerCPI2.0 as a core tool for the concept, which demonstrates generalization ability across proteins and compounds.
Drug resistance mutation analysis
Genome-wide association studies and candidate gene findings suggest that genetic approaches may help in choosing the most appropriate opioid and its dosage, while preventing adverse drug reactions [78]. The analysis of single-nucleotide polymorphisms (SNPs) is rapidly growing, in particular for genetic variants that alter the activity of drug metabolizing enzymes and drug transporters. The 3D structures of proteins are resolved by X-ray crystallography, NMR spectroscopy and more recently by the modern cryogenic electron microscopy, and collected in the Protein Data Bank (PDB). However, some of them are in complexes with ligands and this information is more valuable. Screening of databases, virtually or by high throughput (HTS) assays, is another way to discover new drugs [20,21,22].
Synergy Between the Two Approaches¶
We emphasize that these two analysis methods serve only as interpretation tools, and the systematical evaluation of the prediction of binding sites and activity cliffs is beyond the scope of this work. Here, we propose a sequence-to-drug concept that discovers modulators directly from protein sequences without intermediate steps, using end-to-end differentiable learning (Fig. 1a). End-to-end differentiable deep learning has revolutionized computer vision and speech recognition17 by replacing all components of complex pipelines with differentiable primitives, enabling joint optimization from input to output18. The success of AlphaFold6 in protein structure prediction also relies heavily on the idea of end-to-end differentiability. This concept is appealing because it performs the entire learning process in a self-consistent and data-efficient manner, potentially avoiding the error accumulation of complex pipelines. Many dysregulated pathways are now characterized, and new targets, proteins, and polynucleotides are attracting medicinal chemists.
Pharmaceutical Sciences (PSCI)
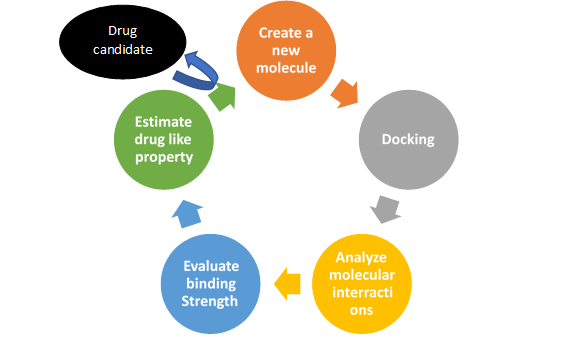
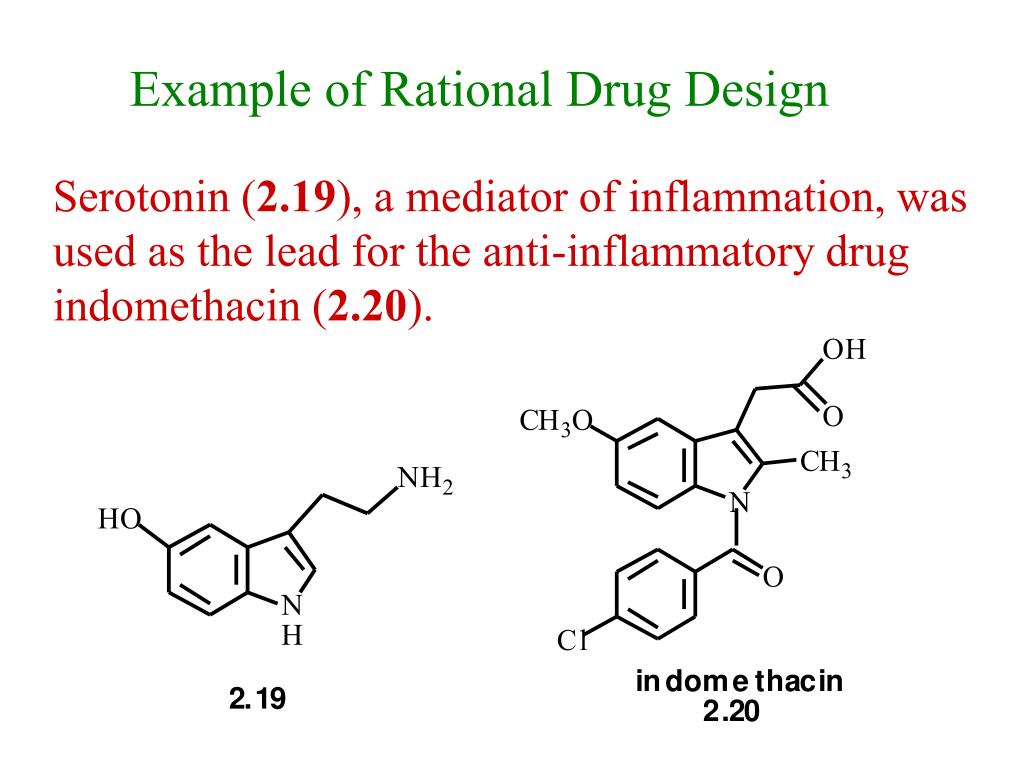
For example, ranitidine is a chemical modification of cimetidine with higher potency and prolonged half-life [18], pindolol originates from propranolol but avoids the first-pass effect in the liver and shows a higher degree of bioavailability [19]. A drug is a foreign molecule that affects biological processes and is used to prevent, diagnose or treat a disease [1]. A molecule can be designed that mimics the structural features common to the set of molecules considered. The ideal situation is to find a way of combining the two approaches; the synergy that is created can substantially accelerate the discovery process. The project can then be intelligently conducted by generating molecules with one approach and by subjecting them to analyses with the second approach. Structure-based design has the advantage of providing a "visual" framework for the direct design of new prototypes.
Finally, we constructed a ChEMBL dataset including 3348 proteins, 69,616 compounds, 117,513 positive CPIs, 134,611 negative CPIs and 252,124 samples in total. According to previous works56,57, we first detected the inhibitory effect of rabeprazole on the activity level of ARF1 in CT26 cells (colon carcinoma cells) using a G-LISA assay. The results showed that rabeprazole effectively inhibited ARF1 activity in CT26 cells in a concentration-dependent manner (Fig. 7a).

PocketFlow is a data-and-knowledge-driven structure-based molecular generative model
These molecules effectively interact with the receptor in a canonical binding mode, while also demonstrating the desired selectivity towards the receptor and favorable ADME properties. To evaluate the potential of DRAGONFLY to generate molecules that extend into new areas of chemical space, we analyzed the similarity of the generated molecules to both the training data set (i.e., a subset of ChEMBL28) and an external data set (i.e., PubChem39, excluding ChEMBL molecules). Our analysis demonstrated that the generated molecules were as similar to the molecules in PubChem when compared to the molecules in ChEMBL (Table S7). While there is a degree of similarity to known molecules, DRAGONFLY also produced a large proportion of molecules with high novelty scores and diverse structures generating higher structural and scaffold novelty than the well-established fine-tuned RNN methods (Table 1). These results suggest that DRAGONFLY is not limited to recapitulating the training data but also has the capacity to explore and generate molecules in previously uncharted regions of chemical space, albeit the extent of this exploration warrants further investigation.
The Master of Science in Drug Discovery and Development is one of over 140 online programs at Drexel University. Through core courses such as Applications of Clinical Research Biostatistics, Current Topics in Pharmacology and Physiology, and Research in Drug Discovery and Development, students will receive comprehensive scientific and technical training. In 1923 the University of Florida established the Master of Science in Pharmacy degree, and in 2000 was able to offer the degree completely online. There are approximately 980 students enrolled in the online courses through the College of Pharmacy. Currently there are two distance learning Pharmacy programs, the 32-credit Master's program in Pharmaceutical Science with a concentration in Pharmaceutical Chemistry, or the 15-credit graduate Certificate.
Upon recruitment of specific cofactors, these heterodimers transactivate PPAR-responsive elements (PPREs) of target genes involved in insulin signaling, lipid and glucose metabolism, immune response, as well as cell cycle and differentiation46,47. Several activators with different selectivity for the respective PPAR subtypes have reached advanced clinical trials or were introduced to the market. Aiming to test the method’s ability to generalize, the training of DRAGONFLY did not include the protein template PPAR or any closely related structures present in the training data set. Specifically, proteins belonging to the same sub-family (PPARα, PPARγ, PPARδ) or other species were intentionally excluded.
It was also reported that the kinase inhibitor gefitinib (Figure 4) and the monoclonal antibody cetuximab share complementary mechanisms of action on EGFR and that a combined EGFR targeting is a clinically exploitable strategy [54]. Computer-aided drug design Computer-aided drug design uses computational chemistry to discover, enhance, or study drugs and related biologically active molecules. The most fundamental goal is to predict whether a given molecule will bind to a target and if so how strongly. Molecular mechanics or molecular dynamics are most often used to predict the conformation of the small molecule and to model conformational changes in the biological target that may occur when the small molecule binds to it. Any advance in science and technology finds immediately its application in medicine, in pharmacy, in drug discovery and development. Investments in drug design are worthwhile because as better is designed a given drug candidate during the experimental stage, as less likely is for the drug to fail in the late stages where the tests are more expensive, especially in the clinical trials.
A shortcut for drug discovery: Novel method predicts on a large scale how small molecules interact with proteins - Phys.org
A shortcut for drug discovery: Novel method predicts on a large scale how small molecules interact with proteins.
Posted: Thu, 25 Apr 2024 18:00:01 GMT [source]
The key advantage of such a method is that novel structures, not contained in any database, can be suggested. Currently, most computational-based methods for identifying drug-target interaction, affinity, and binding sites [4,5,6,7,8,9,10,11,12] utilize deep learning techniques. The general process of these deep learning-based methods involves first collecting a dataset of drug-target interaction. Finally, based on the extracted features, a fully connected (FC) network is used to identify drug-target interaction, affinity, and binding sites. The results of the study also indicate that ligand-based de novo design outperformed structure-based models for the majority of investigated molecular properties.
Revealing Protein-Ligand Interactions Using AI Enables Drug Discovery - Genetic Engineering & Biotechnology News
Revealing Protein-Ligand Interactions Using AI Enables Drug Discovery.
Posted: Fri, 26 Apr 2024 10:33:14 GMT [source]
Many diseases, or at least the symptoms of diseases, arise from an imbalance (either excess or deficiency) of particular chemicals in the body, from the invasion of a foreign organism, or from aberrant cell growth. This approach utilizes a GTNN model to encode the input molecular graph, which is represented as a 2D graph for ligands and a 3D graph for protein binding sites. The GTNN transforms the graph into a condensed one-dimensional (1D) feature vector.
Similarly, the structure-based design DRAGONFLY model encompassed 7.01 million trainable parameters (3.56 million for the GTNN encoder and 3.45 million for the LSTM decoder). Designed and performed the experiments, prepared the figures, and wrote the manuscript; L.C. Contributed to the biological experiments on SPOP inhibitors; H.H., H.G., C. Zhou, Q.S., X. Lu and X.H.
This process involves a complex, human-engineered pipeline with multiple independently optimized steps, and each step has its own limitations5. In particular, the precise predicting active sites remains a challenge as these local structures tend to break the ‘protein-folding rules’9. Another challenge is defining binding pockets for novel targets with multiple domains11, and predicting allosteric sites is still difficult12 due to the varied mechanisms of allosteric effects and high computational costs13. Additionally, structural flexibility allows proteins to adapt to their individual molecular binders and undergo different internal motions9,14,15, making pockets more difficult to define. Finally, virtual screening can generate false positives16 and accumulate errors from the previous two steps.
Given that SPOP is an adapter of E3 ligase and RNF130 is an E3 ligase, the hits we found have the potential to serve as novel warheads for proteolysis-targeting chimaeras (PROTACs). PROTACs have been successfully developed for harnessing the ubiquitin–proteasome system to degrade a protein of interest, receiving tremendous attention as a new and exciting class of therapeutic agents that promise to significantly impact drug discovery. Furthermore, through an inverse application of the sequence-to-drug concept, the FDA-approved drug rabeprazole showed promise for expanding its indications to colon cancer treatment by regulating lipid metabolism and inducing an antitumour immune response. Overall, our findings provide a proof of sequence-to-drug concept, which we believe will become an essential component of future rational drug design pipelines. A The conventional pipeline for target-based drug design and the sequence-to-drug concept. B Three stages of the proof of sequence-to-drug concept, with each stage is labeled by different colors.
"Our work has made the world of proteins accessible for generative AI in drug research," Schneider says. "The new algorithm has enormous potential." This is especially true for all medically relevant proteins in the human body that don't interact with any known chemical compounds. An international, peer-reviewed, open access journal that spans the spectrum of drug design and development through to clinical applications. The journal is characterized by the rapid reporting of application notes, reviews, original research and clinical studies in all therapeutic areas. Clinical outcomes, patient safety, and programs for the development and effective, safe, and sustained use of medicines will be a feature of the journal. The selected hyperparameters for the neural network led to a combined count of trainable parameters amounting to 6.94 million (3.49 million for the GTNN encoder and 3.45 million for the LSTM decoder) for the ligand-based design DRAGONFLY model.








No comments:
Post a Comment